Crucial role of inter-site Hubbard interactions for the correct energetics of spinel Li-ion cathode materials
By Nicola Nosengo, NCCR MARVEL
With demand for electric vehicles and for energy storage expected to grow exponentially over the next decades, the search for more efficient battery materials is in full swing. Both experimental studies and theoretical computations play a key role in this search, and scientists are constantly working to develop better computational tools to explain and predict the behavior of materials that could be used for future Li-ion batteries — in particular, for their cathodes, the pole from which the flow of electrons originate.
In a study recently published in Physical Chemistry Chemical Physics, Iurii Timrov and Michele Kotiuga expanded its use to the study of two candidate cathode materials from the class of lithium-manganese-oxide spinels. The results, which earned the paper the inclusion in the "2023 PCCP HOT Articles" collection, prove that DFT+U+V is indeed a very powerful and accurate method to describe such materials.
Lithium-manganese-oxide spinels once attracted attention as possible cathode materials, because of their low cost, non-toxicity and interesting electrical behavior. But researchers soon found out that, when used in a battery, they quickly show a loss of capacity. That is why they were soon replaced by more efficient materials based on cobalt, such as LiCO2 — which is now used everywhere in portable electronics — olivines, and NMCs — i.e. layered materials composed of nickel, manganese and cobalt that are now used by carmakers. The use of cobalt, unfortunately, poses several problems from unsustainable mining to toxicity, and researchers have not lost interest in the possibility of replacing it with nickel and/or manganese.
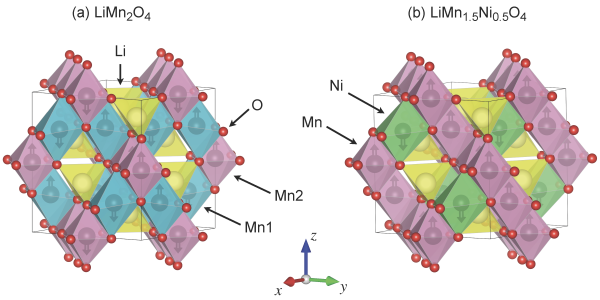
The first material considered by the study is LiMn2O4, and it has been extensively studied for several decades, both experimentally and theoretically, which is why Timrov and Kotiuga chose it as a benchmark to test the accuracy of their method. The second material is a variation on the theme, LiMn1.5Ni0.5O4. "Replacing a fraction of manganese with nickel improves drastically the electrochemical properties of this spinel cathode material" says Timrov. Its unusual composition, however, makes it very difficult to model with existing first-principles methods. In fact, the two scientists note, there are numerous experimental studies on it, but computational ones are almost absent. "It's important for experimentalists to have a better understanding of what they are seeing thanks to the valuable information from simulations", says Timrov. This is what the two authors tried to do with their advanced DFT+U+V method.
DFT+U+V was introduced to better describe the electronic structure of materials in which electrons are not completely localized on atomic sites but are also hybridized with states of the ligands (nearest neighbors). In DFT with standard functionals (local and semi-local such as LDA and GGA), the d and f electrons are overdelocalized due to large self-interaction errors. To fix this, scientists use the Hubbard U correction that makes electrons more localized — this is the widely used DFT+U method. But that's not the end of the story. In some materials there is covalent bonding, where neighboring orbitals overlap and hybridize, and scientists introduced the V parameter to restore a degree of intersite hybridization. "U and V do two opposite things, and the right balance of the two gives the right description of the complex electronic interactions" explains Timrov. This is particularly relevant for materials containing transition metal elements, like those considered by the study. "Depending on the oxidation state of nickel and manganese, some materials are more covalently bonded than others, and this method gives you the flexibility to capture the right degree of hybridization" Kotiuga explains.
The main problem is how to determine U and V. Researchers so far have mostly used an empirical approach, tweaking the two parameters until the calculations match the experiments. But that is not ideal and prevents the application of this method to materials for which there are no experiments yet. "In our lab we have thus developed a new approach to determine both U and V entirely from first principles, based on density-functional perturbation theory" says Timrov.
For both LiMn2O4 and LiMn1.5Ni0.5O4, the method accurately predicts the geometrical properties, with results in remarkable agreement with the experiments. The same is true for oxidation states, band gaps, and for the voltages of batteries based on the two materials, where the results are more accurate than those obtained by other state-of-the-art methods.
The study was performed on the powerful supercomputers of the Swiss National Supercomputing Centre (CSCS) in Lugano. Kotiuga and Timrov explain that DFT+U +V by itself is not hugely expensive in computational terms, but determining U and V from first principles is. Hence there are ongoing efforts in the Marzari lab to speed up the calculations of U and V using machine learning.
As for the future, the authors say that an important next step would be to model the real LiMn1.5Ni0.5O4, instead of the idealized one. This material crystallizes in a slightly different form than the one described by the formula, with a bit less nickel and a bit more manganese, and this affects its behaviour in the real world. More generally, the group will work on other cathode materials, and is working on a high-throughput study on a large database of 3D materials containing lithium. "Now that we have proved the accuracy of the method for certain classes of cathode materials, the idea is to see if we can find promising novel Li-ion battery materials for commercialization" Timrov explains.
Reference
I. Timrov, M. Kotiuga, N. Marzari. Unraveling the effects of inter-site Hubbard interactions in spinel Li-ion cathode materials. Physical Chemistry Chemical Physics 25, 9061 (2023). DOI:10.1039/D3CP00419H
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter