Novel quantum simulation method clarifies correlated properties of complex material 1T–TaS2
by Carey Sargent, EPFL, NCCR MARVEL
1T–TaS2 is a layered transition metal dichalcogenide that has been studied intensively for decades because of intriguing links between temperature dependent distortions in the lattice and phenomena linked to electronic correlations.
Upon cooling, the material undergoes a series of lattice rearrangements with a simultaneous redistribution of the electronic density, a phenomenon known as charge density wave (CDW) order. In the phase reached when the material is cooled to below 180 K, an in-plane periodic lattice distortion leads to the formation of star-of-David (SOD) clusters made of 13 tantalum atoms. Simultaneously, a strong increase in resistivity is observed. Additional interesting properties of the low temperature phase include a transition to a superconducting state under pressure as well as the possibility to switch this phase into long-lived metallic metastable phases by applying short pulses of laser or voltage, making the material potentially interesting for use in future memory devices.
For many years, 1T–TaS2 was considered to be a Mott insulator, and in fact one of the prototypical examples of a single-band Mott system. Ten years ago, theoretical investigations of the electronic structure of 1T–TaS2 proposed a scenario in which a correlation-driven Mott insulating state was formed within the planes but, due to the strong hopping between the layers, a metallic band was present in the stacking direction. Within this scenario, a possible explanation for the insulating nature of the material is stacking disorder, an effect known to exist in the material.
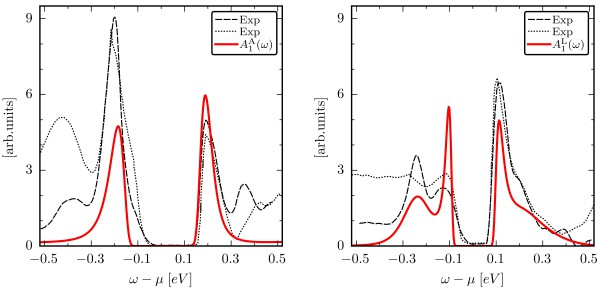
Figure 1. Comparison between the theoretical spectral function (red line) of the surface layer and the recent measurements from "Mottness versus unit-cell doubling as the driver of the insulating state in 1T–TaS2" and "Distinguishing a Mott Insulator from a Trivial Insulator with Atomic Adsorbates" (black lines) for the A (a) and L (b) termination.
Ensuing theoretical investigations into the role of the layer stacking and its effects on the electronic ground state showed that the lowest energy structure exhibits a specific “AL” stacking of bilayers, where A refers to the center of the star-of-David and L to the upper right corner. These results, again not strictly relying on Mott physics, indicate that the insulating state might be due to bonding-antibonding gaps. While this picture can be appropriate for the bulk, the neglect of interaction effects would imply a metallic state pinned to the surface of samples terminating with a broken bilayer, a feature that has been clearly ruled out by several recent scanning tunneling spectroscopy (STS) experiments which systematically reported gapped spectra for both terminations.
This contradiction between theory and experiment prompted researchers at the University of Fribourg to undertake a systematic study of the correlated electronic structure in the stacked bilayer system, using an advanced computational machinery developed within MARVEL. The results of their investigations were recently published in the Physical Review Letters paper Mott versus Hybridization Gap in the Low-Temperature Phase of 1T−TaS2.
The electronic behavior in strongly correlated quantum materials such as 1T–TaS2 cannot be properly described in terms of band structure calculations—theoretical models meant to model such materials accurately must include the effects of strong electronic correlation. The GW + EDMFT ab initio approach for correlated materials modelling, is currently one of the most sophisticated methods available for correlated electron calculations. It has been shown to enable parameter-free simulations of correlated materials. In the present approach, however, the parameters of a multi-layer model were determined by comparison to the known STS spectra for mono-layers. Applying this technique then allowed the researchers to simulate semi-infinite systems of 1T–TaS2 layers in the AL stacking arrangement, identified as the structural ground state in earlier research, for the two different surface terminations.
The calculations performed by postdoc Francesco Petocchi reproduced the spectral features reported in the literature and provided a natural interpretation for the distribution of multiplets observed in photoemission experiments performed by the group of Prof. Claude Monney at the University of Fribourg. Based on their model, they were able to conclude that the insulating behavior of 1T–TaS2 stems from the complex interplay between bonding-antibonding splittings and electronic correlation.
These results, which provide a solid basis for the previous interpretations of recent measurements, indicate that while the bulk region of 1T–TaS2 is essentially a band insulator in the low-temperature phase, the surface region exhibits a nontrivial interplay between band insulating and Mott insulating behavior.
Reference:
Francesco Petocchi, Christopher W. Nicholson, Bjoern Salzmann, Diego Pasquier, Oleg V. Yazyev, Claude Monney, and Philipp Werner. Mott versus Hybridization Gap in the Low-Temperature Phase of 1T−TaS2. Physical Review Letters 129, 016402 (2022)
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter