Ab initio model of Ca2RuO4 perovskite in remarkably good agreement with available experimental data
by Carey Sargent, EPFL, NCCR MARVEL
Electrons in correlated materials experience a strong entanglement between charge, orbital and spin degrees of freedom and this may result in unusual macroscopic phases. Ranging from high-temperature superconductivity to metal-insulator transitions, colossal magnetoresistance and multiferroicity, many such properties could prove useful in new technologies.
Theoretical insights into these materials have been hampered though by a lack of quantitative methods that can accurately describe the strong electron-electron interaction responsible for their unique properties: reliable ab initio description of strongly correlated materials is a long-sought capability in condensed matter physics. Density functional theory (DFT), for example, does not provide an adequate description of the electronic structure of these materials because the approximation of effectively independent electrons breaks down.
Progress in simulating these types of materials has nonetheless been made by supplementing DFT with the missing correlations generated by a dynamical mean-field theory (DMFT) construction. DMFT treats strong local interactions by mapping a many-body lattice problem to a many-body local problem, which is more amenable to numerical treatments. By using the so-called Extended DMFT (EDMFT) it is also possible to capture the effect of nonlocal interactions.
The DFT+DMFT scheme nonetheless has several drawbacks that prevents true ab initio predictions. Such procedures should, for example, provide the interaction parameters that go into the DMFT calculation and they should be consistent with the correlated electronic structure. A more advanced formalism combines the extended (E)DMFT model with the so-called GW method, which allows to incorporate nonlocal self-energy and polarizations into the simulation. This GW + EDMFT approach involves a self-consistent description of correlations and screening in a solid where EDMFT can be combined with the GW method in a consistent way. The result is a true ab initio approach for strongly correlated materials, which does not require user-provided parameters apart from the choice of a subspace of strongly correlated orbitals.
First applications of this recently developed simulation framework have produced promising results, but its reliability has not been systematically tested yet. The question of whether or not the self-consistently computed effective local interaction parameters correctly capture the correlation strength in the material is a particularly pertinent.
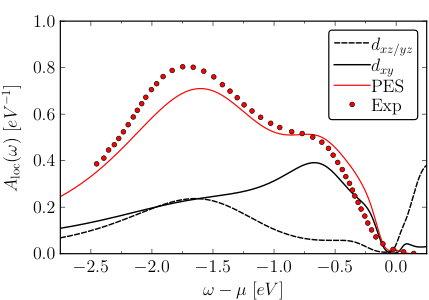
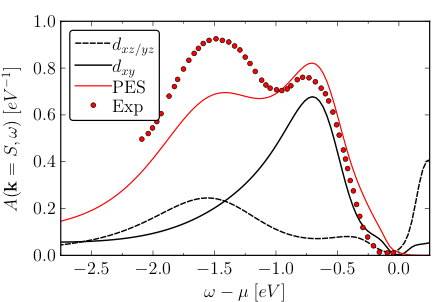
Local and k-dependent spectral function of Ca2RuO4 in the low-temperature phase. The GW+EDMFT prediction (red line) is compared to photoemission data (red dots).
In the paper Fully ab initio electronic structure of Ca2RuO4, recently published in Physical Review B, Dr. Francesco Petocchi, PhD student Viktor Christiansson, and Prof. Philipp Werner at the University of Fribourg tested the reliability of the approach and evaluated its ability to capture the correlation strength by applying it to the compound Ca2RuO4. The material is an experimentally well characterized perovskite compound in which a temperature-dependent structural deformation drives a paramagnetic metal-insulator transition that is not linked to magnetic ordering.
The researchers found that their ab initio approach predicts the physically correct solutions for the two crystal structures and electronic properties in remarkably good agreement with the available experimental data. An EDMFT treatment on the other hand predicts insulating solutions with a far too large gap for both structures: this demonstrates that the nonlocal polarization and self-energy components introduced by GW are essential for setting the correct balance between interactions and bandwidths.
Though a similar level of agreement has been obtained in previous LDA + DMFT studies, these previous calculations required several freely adjustable parameters allowing for the fine-tuning of the simulation results to the experimental reference data. With GW + EDMFT, the user only needs to select the strongly correlated subspace (in this case the three t2g orbitals of Ru). All of the remaining calculations are performed in a fully ab initio manner.
“The level of accuracy demonstrated in this work is therefore a remarkable outcome and a very encouraging result for the further application and development of the GW + EDMFT framework,” the researchers conclude in the paper.
Reference:
F. Petocchi, V. Christiansson, P. Werner, Fully ab initio electronic structure of Ca2RuO4, Phys. Rev. B 104, 195146 (2021)
DOI: 10.1103/PhysRevB.104.195146
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter