New algorithm allows for the rapid identification of entire reaction pathways in complex systems
By Carey Sargent, NCCR MARVEL, EPFL
Determining a reaction or transformation pathway is critically important in fields including chemistry, physics and materials sciences. Researchers have correspondingly proposed numerous methods of doing so.
Many require atomic index mapping as input: it is essential to know which atom in the reactant is mapped onto which atom in the product. The problem is that atoms are indistinguishable—one carbon atom, for example, is the same as any other. In a chemical reaction, any one atom of a given type in the reactant can, in principle, be mapped on to any atom of the same type in the product. While it can be easy to track atoms in simple reactions, the problem becomes difficult in more complicated cases.
While complex reaction and transformation pathways involving many intermediate states could in principle be solved on the density-functional theory level with existing simulation methods, it is generally impossible because of the huge number of energy and force evaluations required. If the chemical reaction system consists of M1 atoms of type 1, M2 atoms of type 2, there are M1!M2!;… different possible mappings.
“N factorial is a function that increases extremely rapidly and so in practice you can never try out all the possible mappings,” said Stefan Goedecker, Professor of Physics at the University of Basel. “We cannot determine which atom in the reactant is mapped onto which atom in the product.”
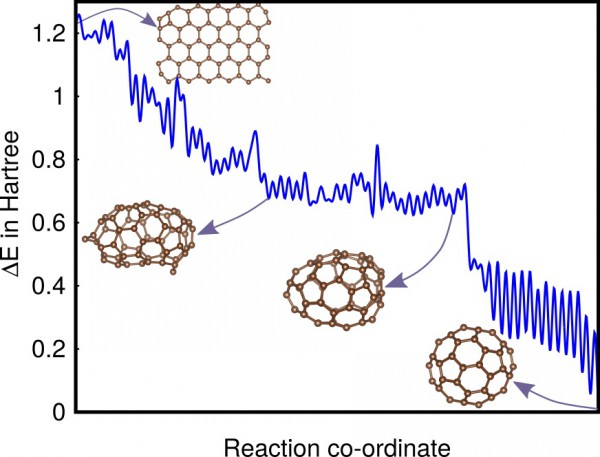
Typical low-barrier pathway of C60 from a graphene
flake to a fullerene.
Though alternative approaches such as the so-called one-sided methods, molecular dynamics techniques, metadynamics and temperature accelerated dynamics exist, they are linked to other complications. This inspired the researchers to develop a new technique based on an approach Goedecker himself first developed in 2004, the minima-hopping method.
This simulation approach is used to search for the global minimum of the potential energy surface of complex molecular systems. Finding this global minimum is critical in numerous fields because it gives the crystalline ground state structure of a solid for periodic systems and determines the geometric ground state configuration of molecules for nonperiodic systems, for instance. The innovation of the minima-hopping method was to explore the configurational space as fast as possible and avoid revisiting already known parts. It also incorporated a preference towards low energy configurations.
“In principle, global optimization is also part of that class of problems that is not solvable, but it turns out that if the function you want to optimize has certain beneficial properties, it can be done very easily.”
--Stefan Goedecker
Now, the researchers in Basel have added to this basis a function that drives the simulation towards the desired final configuration, or reaction product. Based on configurational distances derived from fingerprints that remain the same under atomic index permutations—meaning that it is not necessary to say which atom is mapped to which other atom at any point in the algorithm—the penalty function can uniquely identify a single configuration. The fingerprint distance is zero only if the configuration is exactly identical to the desired final configuration. This biases the potential energy surface so that it is essentially a “structure seeker” whose global minimum is the reaction product.
“Our approach, instead of dealing with this combinatorial problem, was to map it onto a global optimization problem,” Goedecker said. “In principle, global optimization is also part of that class of problems that is not solvable, but it turns out that if the function you want to optimize has certain beneficial properties, it can be done very easily.”
Research in the paper concentrated on complex reaction pathways where the system had to cross a number of transition states, the highest energy points on the lowest energy pathway connecting a chemical reactant to a product. Starting from an initial configuration, or reactant, the researchers “travelled” through the minima hopping method’s consecutive intermediate states until arriving at the ground state.

What’s more, since the hops from one local minimum to the next in the minima hopping method are based on molecular dynamics followed by local geometry optimizations, the identity of individual atoms of the same type can be traced back for any hop. This results in the correct mapping of the atomic indices for the entire complex reaction pathway.
“Density function calculations are expensive and if you had to search for a reaction pathway on an unbiased potential energy surface it would take forever, there’s no chance that you could do it,” Goedecker said. “Now we’ve introduced this bias and our potential energy surface has these nice features and we can fall down very rapidly into the global minimum. This path is the reaction pathway.”
The scientists first demonstrated that the method finds intermediate states relevant to the lowest energy reaction pathway for a benchmark system. They then applied the method to two real systems, C60 and C20H20, and showed that the reaction pathways found contain valuable information on how these molecules can be synthesized.
The results indicate that the method gives insight at the atomistic level into complex reaction pathways such as those found in catalysis, as well as complex phase and shape transformations in nanoparticles. It could also help with the discovery of new organic-chemistry synthesis pathways.
Reference: Finding Reaction Pathways with Optimal Atomic Index Mappings
Deb Sankar De, Marco Krummenacher, Bastian Schaefer, and Stefan Goedecker
Phys. Rev. Lett. 123, 206102 – Published 15 November 2019
DOI: 10.1103/PhysRevLett.123.206102
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter