Machine learning presents a huge opportunity to identify new materials at a reduced cost
Carey Sargent, EPFL, NCCR MARVEL
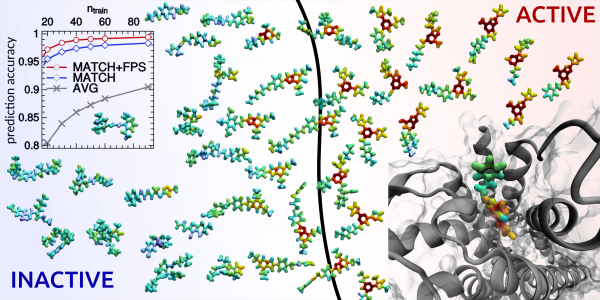
A machine-learning algorithm can identify active and inactive protein ligands with 99% accuracy using only 50 training points. The same scheme can be used to build potentials for condensed-phase applications and to predict with unprecedented accuracy the properties of small organic molecules.
Credits: Michele Ceriotti, EPFL and co-authors.
To achieve the feat, scientists in the group of Anatole von Lilienfeld, professor and head of the QM-RCD Lab at the University of Basel and a member of MARVEL, first used density functional theory to generate training data on the predicted formation energies of two sets of possible elpasolites, crystals principally made of aluminum, fluorine, potassium and sodium and used in the fabrication of scintillators. The first, smaller set predicted the energies for crystals variants that might also include 12 selected elements including carbon and nitrogen while the second, larger set included possible combinations with all main group elements up to bismuth. The first training data set included a complete list of formation energies while the second, much larger set, was generated through the random selection of possible crystals.
After testing the performance of the ML model and showing that it can be generalized to address the properties of any crystalline material, the scientists used it to investigate possible elpasolites. They calculated the formation energies of two million crystals and were able to identify 90 that are thermodynamically stable, some of which have interesting electrical properties. The team calculated the crystals with a single laptop in one afternoon, bringing the cost of computing time from between CHF 0.5 million and CHF 2 million to essentially nothing.
“The advantage of these techniques is basically that you dramatically speed up the time you need to study any given new material by multiple orders of magnitude,” von Lilienfeld said. “Whenever you have a speed-up or improvement on the order of multiple orders of magnitude, typically disruptive, revolutionary things happen. “

Michele Ceriotti (left), Anatole von Lilienfeld (middle) and Martin Jaggi (right)
Studying properties linked to the behavior and interaction of electrons and nuclei is a critical part of the work done at NCCR-MARVEL. This involves sophisticated levels of theory and therefore significant investment in terms of CPU time. MARVEL has the distinct advantage of working with three professors—von Lilienfeld in Basel and Michele Ceriotti and Martin Jaggi at EPFL— who specialize in machine learning techniques. Their approaches allow researchers to study a set of materials computationally and then use this data to make educated guesses about new materials. If the training data set is large enough and sufficiently well-chosen, a critical point, predictions about the new material should be as accurate as those generated by conventional methods. The approaches can save time and therefore money, but also identify potential molecules that might not otherwise be conceived. This is the promise of machine learning approaches in material science.
The example from von Lilienfeld’s lab is just one example of how the three professors focused on these techniques in MARVEL are turning to machine learning approaches to discover and study new materials. Researchers in the group of Michele Ceriotti, professor and head of the Lab of Computational Science and Modelling at EPFL, used machine learning techniques to model possible polymers of a molecular crystal that has been used in organic electronics, like OLED.
They were able to show that they can predict the stability and electron mobility of the different structures as well as the energy and charge mobility in the crystals using about one-tenth of the calculation time necessary with the usual techniques. Since determining the stability of molecules and condensed phases is the basis for understanding chemical and materials properties and transformations, the group’s approach effectively provided a unified framework for predicting these atomic scale properties.
“The kind of machine learning we do at MARVEL has to do with circumventing expensive quantum chemistry calculations and predicting the atomic scale properties of materials using just a small number of reference calculations,” Ceriotti said. “Rather than doing thousands or millions of density function or whatever kind of calculations, you run a few hundred carefully selected ones and then use statistical regression to predict the behavior of other molecules that are similar enough to the one that you have computed that you can effectively interpolate between them.”
“Effectively” is indeed a critical notion. Ceriotti’s group’s approach predicted the stability of different classes of molecules with chemical accuracy and distinguished active and inactive protein ligands with more than 99% reliability. The model from von Lilienfeld’s group achieved the same, or better, accuracy with respect to the classical method of density functional theory in comparison to experimental data.
The methods also continue to improve in predictive power as the size of the data set increases, von Lilienfeld said: converging to arbitrary accuracy is really possible. This is a stark difference from the conventional regression method that people have been using in the engineering sciences for decades, and the underlying reason for the hype surrounding big data: these methods allow researchers to really make use of it.
It’s similar, he said, to the predictive power of a machine learning algorithm when presenting new movies or books to users of Netflix or Amazon. The more active the user, the more accurate the prediction, and the more likely the marketing and ad placement for this user will be successful.
“We have succeeded in doing something similar with materials,” he said. “The more data you have on different materials, the more materials you study, the more accurate you’ll be when you train the machine learning model on it.”
Despite clear advantages of the machine learning approaches, the researchers warn that they are not silver bullets that can solve anything. There are important scientific, industrial or technological problems that are not suited to machine learning approaches.
Suitable projects generally have to meet a set of requirements, von Lilienfeld said. The idea must be to quantitatively predict a property in a high dimensional space; it must be possible to obtain reasonable amount of data within a reasonable time or money budget¸ and researchers should have a certain degree of understanding as to how the function is related to the input.
Martin Jaggi, professor and head of the Machine Learning and Optimization Laboratory at EPFL, confirms the point, saying that potential collaborators sometimes think that they can simply show up with a big hard drive full of data.
“Training data is not just data, it’s input data together with a useful training answer, a property to predict” he said. “People often forget about that part.”
Jaggi, whose group works on large-scale machine learning as well as text understanding, says the most successful collaborative projects require considerable amount of domain knowledge including a good understanding of what information might be in the data.
“If all of this is there, the project can be really interesting,” he said.
On the same topic : Can artificial intelligence create the next wonder material? Nature, May 2016, Nicola Nosengo.
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter