Tackling excited states: Koopmans functionals now available as an open-source software package
By Nicola Nosengo, NCCR MARVEL
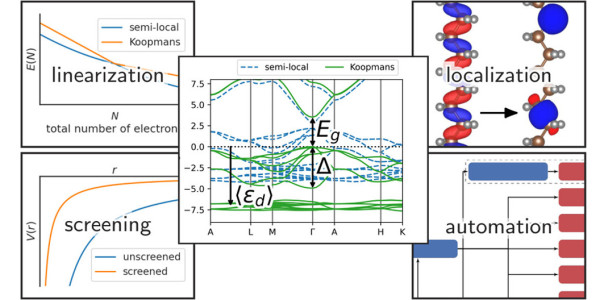
Density functional theory (DFT) is a cornerstone of materials simulation, but it has well known limitations. It allows scientists to calculate the behaviour of electrons inside a material in their lowest energy configuration, but it fails to accurately describe excited states, and thus cannot be used to describe the spectral properties of molecules and materials – such as the energy of light required to eject an electron from a material in a solar cell. Scientists use other computational techniques to study these processes, such as the GW method, but they are computationally more expensive, require a lot of fine-tuning, and are not easy to deploy on a large scale.
That is why, about a decade ago, researchers in Nicola Marzari’s group at EPFL embarked on an effort to devise a new set of functionals that would enable DFT to reliably calculate spectral properties. They named their new functionals “Koopmans” functionals, after the Dutch mathematician and economist Tjalling Koopmans, author of a foundational theorem on the ionization energy of molecular systems (and a Nobel laureate in economics to boot)
These Koopmans functionals showed excellent promise; they predicted excited state properties well, and the researchers continued to develop them, but there was one major problem.“The very early implementations of these functionals were very inaccessible, private codes” recalls Edward Linscott, a scientist in Nicola Marzari’s laboratory at EPFL and lead author of the new study. “After all, the theory was very novel, there was a lot of unexplored territory, and the formulas were quite different to what existing codes implemented. But over time, we refined our computational techniques and our code base matured, culminating in the release of a code that now can be used also by non-experts”.
J. Chem. Theory Comput. 2023.
Reference
E. B. Linscott, N. Colonna, R. De Gennaro, N. Linh Nguyen, G. Borghi, A. Ferretti, I. Dabo, and N. Marzari, "koopmans: An Open-Source Package for Accurately and Efficiently Predicting Spectral Properties with Koopmans Functionals". Journal of Chemical Theory and Computation (2023). DOI: 10.1021/acs.jctc.3c00652
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter