DFT+U+V for accurate electronic properties of olivine-type Li-ion cathode materials
By Carey Sargent, EPFL, NCCR MARVEL
Increasing need for renewable energy and energy storage technology has led to considerable research in many areas—the development of lithium-ion (Li-ion) rechargeable batteries has been one major advance.
Already used in applications ranging from portable electronics to the automotive industry and electricity grids, demand is increasing on the back of a global increase in energy consumption and increasing dependence on these efficient, safe, and nontoxic batteries.
The properties and performance of the batteries in terms of power and energy density, capacity retention, cyclability and thermal stability depend on many factors and how the battery components interact. Cathode materials play a pivotal role in this, determining the Li intercalation voltage and cyclability of Li+ ions through the interface with the electrolyte.
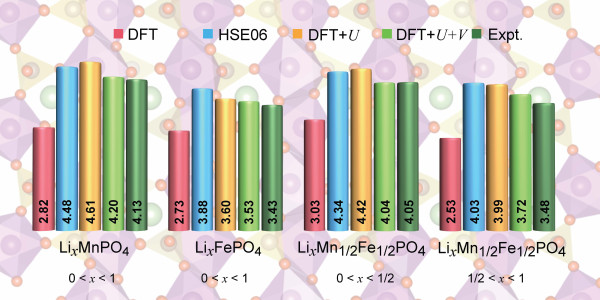
Transition metal (TM) elements—electrochemically active species that change their oxidation state during charging and discharging of the battery—are key ingredients of these cathodes. Understanding at the atomistic level how the properties of these compounds lead to efficient electrochemical processes is therefore critically important. While density-functional theory (DFT) calculations with local and semilocal exchange-correlation functionals can play an important role in producing first-principles predictions for these materials, they can also produce unsatisfactory results in terms of data generated on, e.g., voltages and formation energies. This is due to self-interaction errors that are particularly large for partially-filled d states of TM elements when using standard local and semilocal exchange-correlation functionals.
Researchers have therefore put much effort into developing novel functionals that are more accurate than standard ones to characterize these materials. It is still necessary, however, to determine which functional gives the the most accurate, reliable, and computationally affordable results for the properties of cathode materials. In the paper “Accurate Electronic Properties and Intercalation Voltages of Olivine-Type Li-Ion Cathode Materials from Extended Hubbard Functionals,” recently published in Physical Review X Energy, researchers led by NCCR MARVEL’s Iurii Timrov and Nicola Marzari present a detailed comparative study of four electronic-structure methods for selected olivine-type cathode materials: LixFePO4, LixMnPO4, as well as the more complex mixed-TM compound LixMn1/2 Fe1/2PO4 (x = 0, 1/4, 1/2, 3/4, 1).
After performing calculations with the four electronic-structure methods DFT, DFT+U, DFT+U+V, and HSE06, they determined that the DFT+U+V method clearly outperforms the other well-established methods in terms of reliability of their predictions of features such as oxidation states and Li intercalation voltages, as compared with experiment.
In DFT+U+V, U is a numerical onsite Hubbard parameter meant to fix interactions between electrons on the same atom, while V represents the strength of the intersite interactions between electrons on neighboring sites. In the paper, U and V were computed from first principles using density-functional perturbation theory and did not require any adjustments or ad hoc fitting of the model.
The researchers showed that the DFT+U+V approach displays very clearly “digital” changes in oxidation states of the transition-metal ions in all compounds, including the mixed-valence phases occurring at intermediate Li concentrations. This leads to voltages in remarkable agreement with experiments.
They also showed that including intersite Hubbard interactions is essential for accurately predicting thermodynamic quantities because it balances the drive for localization induced by the onsite U with intersite V orbital hybridizations. This allows DFT+U+V, in contrast to other methods, to accurately describe such localization-hybridization interplay. In doing so, it “opens the door for the study of more complex cathode materials as well as for a reliable exploration of the chemical space of compounds for Li-ion batteries,” the researchers said.
References:
I. Timrov, F. Aquilante, M. Cococcioni, N. Marzari, Accurate Electronic Properties and Intercalation Voltages of Olivine-Type Li-Ion Cathode Materials from Extended Hubbard Functionals, PRXEnergy, 1, 033003, 2022
DOI: https://doi.org/10.1103/PRXEnergy.1.033003
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter