Polarons free from many-body self-interaction in density functional theory
by Carey Sargent, EPFL, NCCR MARVEL
Evidence of polarons—units formed by an electron or hole and the self-generated potential well it sits in—have been found in many materials, including transition-metal oxides, organic semiconductors, conducting polymers and 2D materials. Polaronic states affect the energetic and transport properties of electrons and holes, and related effects have been identified as critical factors in phenomena such as charge transport, surface reactivity and thermoelectricity. Accordingly, scientists in the physics, chemistry, and material science communities have shown great interest in these quasiparticles.
Polarons create potential wells by displacing surrounding ions, creating a cloud that follows the charge carrier as it moves through a material. The strength of the electron-phonon interaction determines its spatial extent and varying nature and properties: with weak coupling, the polaron has a large spatial extension; with strong coupling, the polaron localizes over a short length scale comparable to the lattice parameter. They are, accordingly, referred to as large and small polarons, respectively.
While large polarons are generally studied through Monte Carlo methods, researchers typically use first-principles approaches based on density functional theory (DFT) to investigate small polarons. DFT is a powerful tool for modelling the atomic and electronic structures of materials, but it meets its limits in describing small polarons because it does not correctly account for electron self-interaction (SI). This SI affects the polaron energetics and may even oppose the electronic and structural localization of the polaron.
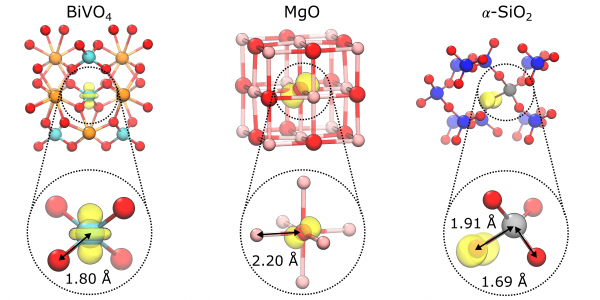
Figure 1: Polaron isodensity surface at 5% of its maximum calculated with PBE0(αk) for the electron polaron in BiVO4, the hole polaron in MgO, and the hole trapped at the Al impurity in α−SiO2 (Bi in orange, V in cyan, O in red, Mg in pink, Si in blue, Al in grey).
Researchers distinguish between two types of SI: one-body and many-body. One-body SI generally refers to how the interaction of a charge with itself is canceled, according to the Hartree-Fock theory, a method of approximation for determining the wave function and the energy of a quantum many-body system. Many-body SI on the other hand refers to the deviation from the piece-wise linearity of the total energy with electron occupation.
Scientists have proposed various approaches to correctly modelling both, but no individual method has proven totally satisfactory. Furthermore, it is not yet clear which SI plays the dominant role in polaron physics and so investigating the two comparatively may help clarify the situation. In the paper “Many-Body Self-Interaction and Polarons,” recently published jointly in Physical Review Letters and in Physical Review B as an Editor’s Suggestion "Polarons free from many-body self-interaction in density functional theory," researchers Stefano Falletta and Professor Alfredo Pasquarello of the Chair of Atomic Scale Simulation at EPFL investigate the concept of many-body self-interaction in relation to polarons in density functional theory. In doing so, they aimed to develop a comprehensive understanding of the relationship between one-body and many-body SI, but also to develop an approach to modelling them using semi-local DFT, which could facilitate wider study of polarons.
The researchers started out by developing a unified theoretical formulation that encompasses both many-body and one-body forms of SI. They find an analytical formulation for both many-body and one-body SI and show that they are related through the dielectric constant. They demonstrate that the two forms coincide when there is no electron screening, indicating that the many-body form of self-interaction is preeminent.
This observation then allowed them to develop a formulation for treating both electron and hole polarons at the semilocal level of DFT. They showed, using examples of three different polarons—the electron polaron in BiVO4 , the hole polaron in MgO, and the hole trapped at the Al impurity in α-SiO2 – that the approach yields polarons with charge densities, atomic structures, and formation energies in close agreement to those obtained from a much more computationally expensive approach. The result indicates the robustness of the electronic and structural properties of polarons free from many-body self-interaction, the authors said.
“Our paper advances the conceptual understanding of the self-interaction problem in density functional theory,” the authors said. “This paves the way to efficient calculations of polarons in large systems, in systematic studies involving large sets of materials, and in molecular dynamics evolving over long time periods.”
References: S. Falletta and A. Pasquarello, Polarons free from many-body self-interaction in density functional theory, Phys. Rev. B 106, 125119 (2022).
https://doi.org/10.1103/PhysRevB.106.125119
S. Falletta and A Pasquarello, Many-Body Self-Interaction and Polarons, Phys. Rev. Lett. 129, 126401 (2022).
https://doi.org/10.1103/PhysRevLett.129.126401
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter