Automated Wannierisation for high-throughput computational materials design
by Carey Sargent, NCCR MARVEL, EPFL
The combination of modern high-performance computing, robust and scalable software for first-principles electronic structure calculations as well as emerging computational workflow management platforms is accelerating the design and discovery of materials with tailored properties through first-principles high-throughput (HT) calculations.
Wannier functions (WFs) play a key role in these electronic structure calculations for two main reasons. They can bridge length scales by allowing for the transfer of information such as density-functional theory calculations from the atomic scale to mesoscopic scales at the level of functional nano-devices. Compact WF representations also provide a means of computing, at much lower cost but without loss of accuracy, advanced materials properties that require very fine sampling of electronic states in the Brillouin zone (BZ).

Figure 1: High-throughput in automated Wannierisation
Among these, the so-called maximally-localized Wannier functions (MLWFs) are the most widely used in actual calculations in the solid state. An essential element of the related minimization procedure is the specification of a set of initial guesses for the MLWFs. These are typically trial functions localized in real space and are specified by the users, based on experience and chemical intuition. In the case of an isolated manifold of bands, the final result for the MLWFs is almost always independent of the initial guess. In cases with entangled bands however, the initial guess strongly affects the quality of the final MLWFs—this complicates the development of a general-purpose approach to generating MLWFs automatically, without user intervention. Several approaches to automating the generation of MLWFs have been proposed.
One such method is a recently proposed algorithm by Damle et al. known as the “selected columns of the density matrix” (SCDM) method, that has shown great promise in avoiding the need for user intervention in obtaining MLWFs. Based on linear algebra techniques for decomposing matrices rather than chemical intuition, SCDM is robust and can be used without the need for an initial guess, making it well suited to HT calculations. Damle and colleagues also proposed an efficient algorithm for a given factorization that operates on a smaller and numerically more tractable matrix than the full density matrix. Finally, the approach allows for parameter-free Wannierisation for an isolated set of composite bands, and only requires three parameters—the number of Wannier functions N and two real numbers μ and σ —in the case of entangled bands.

In the paper “Automated high-throughput Wannierisation,” [1] researchers propose a fully automated protocol—also in the case of entangled bands—for constructing MLWFs based on the SCDM approach. Because SCDM can be used to automatically choose the initial subspace, the approach allows them to avoid the high sensitivity of iterative minimization algorithms to the initial conditions.
The appropriate choice of the two real numbers for use in cases of entangled bands is critical for the success of the method, as is the number of Wannier functions of the manifold to be described. The researchers addressed these issues with a protocol that choses these parameters by using information that is encoded in the projectability of the Bloch states on pseudo-atomic orbitals.

They demonstrated the robustness of the approach by carrying out high-throughput calculations on a dataset of 200 bulk crystalline materials, 81 of which were insulators, spanning a wide chemical and structural space. The main metric used to assess the results was the so-called band distance, which quantifies the difference between the Wannier-interpolated band structures and the corresponding direct DFT band structures. They generally obtained excellent interpolations: for entangled bands, 97% of the materials show an average band distance of less than 20 meV, and a full 72% show a distance of less than 5 meV. For insulators, 93% show a band distance of less than 2 meV when limiting the investigation to valence bands only. In general, they found that the SCDM method works very well for band-structure interpolations, but does not always perform as well for other kind of applications where, for instance, a specific symmetry character of the WFs is desirable.
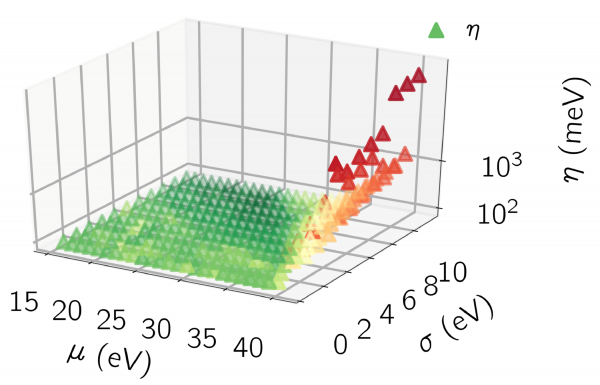
Figure 2: Total position spread Ω (right panel) using SCDM+MLWF for tungsten(W), as a function of the SCDM input parameters μ and σ.
To make the method available to any researcher, they implemented the SCDM algorithm in pw2wannier90, part of the open-source Quantum ESPRESSO distribution, and added corresponding functionality to the open-source Wannier90 code. The full procedure has also been implemented as AiiDA workflows, encoding the knowledge that is needed to perform all steps including DFT simulations, selection of the parameters and Wannierisation into automated software. Researchers can obtain MLWFs and use them to calculate material properties by providing a material’s crystal structure as the only input.
Taking the interests of Open Science even further, the researchers have also distributed publicly and freely all codes and workflows discussed in the paper through a virtual machine [2] that has been preconfigured with the open source codes AiiDA, Quantum ESPRESSO and Wannier90. This virtual machine allows anyone to explore and easily reproduce their results without needing to install or configure anything, and without having to reimplement workflows and algorithms. Interested researchers do not need to re-run the calculations performed in the paper, but can perform their own simulations, either with different parameters or on new materials.
References:
[1] Vitale, V., Pizzi, G., Marrazzo, A. et al. Automated high-throughput Wannierisation. npj Comput Mater 6, 66 (2020).
https://doi.org/10.1038/s41524-020-0312-y
[2] Vitale, V., Pizzi, G., Marrazzo, A. et al. Automated high-throughput Wannierisation. Materials Cloud Archive 2019.0044/v2 (2019).
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter