Novel MD simulation sheds light on mystery of hydrated electron's structure
by Carey Sargent, NCCR MARVEL, EPFL
The e-aq species is difficult to observe directly because it is short-lived and cannot be separated or concentrated. This rules out using direct structural approaches, diffraction or NMR spectroscopy to explore its structure. Though some properties including spectra in UV and IR-regions and the binding energy have been directly observed, the general lack of direct experimental measurements of the structure of the hydrated electron calls for theory.
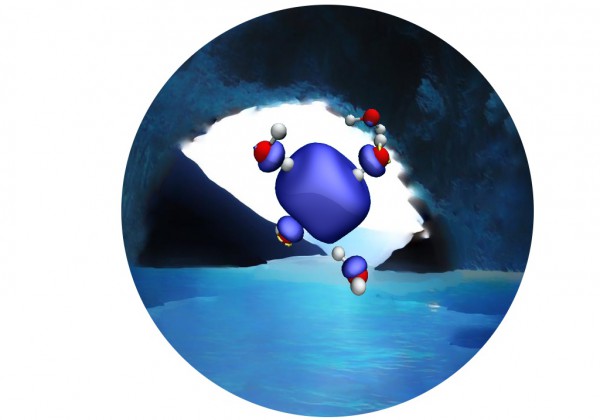
A novel MD simulation provides conclusive evidence in favor of a persistent tetrahedral cavity made up of four water molecules. Credit: Vladimir Rybkin
Reliable modelling of the hydrated electron is at least as challenging as the experimental approach though and the limitations of the computational approaches applied so far have led to considerable theoretical uncertainty. Researchers have not, for example, been able to agree on whether or not the hydrated electron occupies a cavity. Though most theoretical studies suggest that it does, non-cavity models have also proven accurate. Another point of discussion is linked to the distinguishable surface and bulk structures of the hydrated electron.
In the paper, researchers Vladimir Rybkin and Jan Wilhelm at the University of Zurich and Joost VandeVondele at ETH Zurich and CSCS used the first molecular dynamics simulation of the bulk hydrated electron based on correlated wavefunction theory to provide conclusive evidence in favor of a persistent tetrahedral cavity made up of four water molecules. They also showed that there are no stable noncavity structures in the bulk hydrated electron.
The scientists arrived at their model through careful consideration of what features the most accurate approach must have. They wanted it to be based on molecular dynamics to capture the formation and dynamic transformations of the cavity. They needed a many-body correlated electronic structure level to avoid delocalization error and to allow for the correlation effects that have been found crucial in predicting the solvation of the electron accurately without empirical parameters. They wanted the simulation to be performed in the bulk under periodic boundary conditions to avoid formation of the surface structure and, finally, the method should provide an accurate description of liquid water.
The MD simulation they devised--the very first dynamic simulation of a complex chemical species in the condensed phase at the correlated wave function level of theory--fulfills all of these requirements. This was the first critical step. The second was actually managing to carry it out. Such calculations have been technically impossible until recent advances--including those made in their own groups--have allowed for massively parallel manybody theory calculations in the condensed phase on stateart supercomputers such as those as CSCS. It nonetheless took them about 1 million node hours on the Piz Daint supercomputer, Europe's fastest.
The model showed that a cavity is formed within 250 femtoseconds after the excess electron is added to the unperturbed liquid water. Critically, the simulation failed to find any evidence of noncavity structures of the bulk hydrated electron in either stable or metastable states: this provides much stronger theoretical evidence for the cavity model.
The work was funded in part by NCCR MARVEL.
Reference: Dr. Jan Wilhelm, Dr. Joost VandeVondele, Dr. Vladimir V. Rybkin, Dynamics of the Bulk Hydrated Electron from Many-Body Wave-Function Theory, Angewandte Chemie International Edition (2019). DOI: 10.1002/anie.201814053
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter