MARVEL Mini-Series on DFT, wavefunction methods, machine learning and metals
Program in a nutshell
Jan Gerit Brandenburg (University of Göttingen & University College London) — January 24, 17:00, MED 2 1124 (CoViz2), EPFL
Towards the design of molecular materials: from many-body methods to enhanced density functional approximations
Francesco Aquilante (University of Geneva) — January 25, 11:00, MED 2 1124 (CoViz2), EPFL
At odds with electron correlation: are we betting solely on the winning horse?
Volkan Cevher (EPFL) — January 28, 11:00, CO 121, EPFL
Robust and Practical Bayesian Optimization and Beyond
Ralf Drautz (Ruhr-Universität Bochum) — January 30, 11:00, MED 2 1124 (CoViz2), EPFL
Development and validation of interatomic potentials and application to the simulation of phase transformations

From left to right: Jan Gerit Brandenburg, Francesco Aquilante, Volkan Cevher and Ralf Drautz
Abstracts
January 24, 17:00 — MED 2 1124 (CoViz2), EPFL
Towards the design of molecular materials: from many-body methods to enhanced density functional approximations
Jan Gerit Brandenburg (University of Goettingen, Germany & University College London, UK)
New technologies are made possible by new materials, and until recently new materials could only be discovered experimentally. However, approaches based on the fundamental laws of quantum mechanics are now integrated to many design initiatives in academia and industry (see Fig.1), underpinning efforts such as the Materials Genome initiative or the computational crystal structure prediction (CSP [1]). The latest CSP blind test organized by the Cambridge Crystallographic Data Center [2] revealed two major remaining challenges:
(i) Crystal polymorphs are often separated by just a few kJ/mol, exceeding the accuracy of standard density functional approximations (DFAs).
(ii) Dealing with a vast search space, in particular for molecules with increased flexibility, one has to sample about 1 Mio possible crystal structures.
Recent algorithmic developments in Quantum Monte-Carlo make it feasible to molecular crystals and we are now able to predict static lattice energies with potentially sub-chemical accuracy [3]. On the other hand, cost-effective electronic structure methods will be presented that gain up to four orders of magnitude in computational speed compared to traditional DFAs and are suited for optimizing a huge number of putative crystal structures [4]. Promising applications to the CSP of pharmaceutical-like molecules have been demonstrated recently [5]. A perspective on employing machine learning techniques in the CSP context will be discussed.
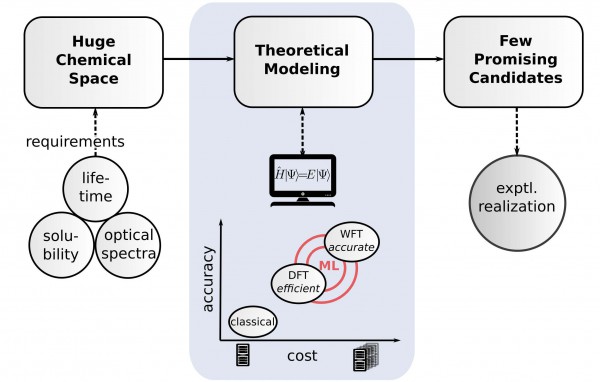
Fig.1. Sketch of a possible materials discovery work-flow. The theoretical modeling connects target properties with the experimental realization by reducing the huge space of potential materials to a few promising ones. The compromise between high accuracy and affordable computational cost in solving the Schrödinger equation is shown
[1] S. L. Price, JGB, Molecular Crystal Structure Prediction; Elsevier Australia, 2017.
[2] A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, JGB, P. J. Bygrave, R. Bylsma, J.E. Campbell, R. Car, et al. Acta. Cryst. B 2016, 72, 439.
[3] A. Zen, JGB, J. Klimeš, A. Tkatchenko, D. Alfè, A. Michaelides, Proc. Natl. Acad. Sci. USA 2018, 115, 1724.
[4] E. Caldeweyher, JGB, J. Phys.: Condens. Matter 2018, 30, 213001.
[5] L. Iuzzolino, P. McCabe, S. L. Price, JGB, Faraday Discuss. 2018, 211, 275.
About the speaker — Following his Diplom in physics at Heidelberg University, Dr. Brandenburg completed his dissertation in Theoretical Chemistry in 2015. He moved to the University College London as a visiting lecturer funded by the Alexander von Humboldt foundation. In 2018, he moved back to Germany, where he currently continues his research at the University of Göttingen. His research involves computer simulations of molecular crystals with specific focus on the prediction of organic crystal structures and their properties. He develops and applies simplified density functional based electronic structure approaches as well as many-body methodologies. Dr. Brandenburg has been awarded numerous early career prices, among them the PhD price of the university society Bonn for the best thesis over all disciplines. His research has been published in over 40 peer-reviewed articles. He is partner of the ERC consortium NanoSolveIT and contributor of an INCITE 2019 project funded by the U.S. Department of Energy.
January 25, 11:00 — MED 2 1124 (CoViz2), EPFL
At odds with electron correlation: are we betting solely on the winning horse?
Francesco Aquilante (University of Geneva)
The theoretical study of realistic systems of interest in life sciences or nanotechnology is possible only if the contrasting goals of high accuracy and low computational costs are met. Despite its success and increased popularity, DFT fails at times in the ultimate goal of being a predictive tool. For this reason, development of alternative electron correlation methods is still a very active area of research in Quantum Chemistry. This talk starts with an overview on some of the computational techniques developed around the paradigm of multiconfigurational WFT. Several key elements are discussed. First, how such methods can be useful for the study of strong correlation in molecules. Second, the way we can build robust multiscale approaches on top of WFT in order to dissect electron correlation effects into components of different characteristic length-scale. The talk will end with a presentation of some ideas aimed at the design of a new class of DFT functionals with the help of information from WFT.
About the speaker — Francesco Aquilante holds a PhD in theoretical chemistry from Lund University with a thesis on the development of the so-called ab initio Density Fitting from Cholesky Decomposition approximation.After a postdoctoral stay at Geneva University, he became an independent researcher at Uppsala University and then obtained a grant from the Italian Research Ministry to carry on his work at the University of Bologna. He is now completing a second research stay at Geneva University. One of the major contributors to the MOLCAS quantum chemistry software, his expertise spans from multiscale techniques for excited state calculations to the treatment of strong correlation in molecules through electron correlation methods and novel density functional approximations.
January 28, 11:00 — CO 121, EPFL
Robust and Practical Bayesian Optimization and Beyond
Volkan Cevher (EPFL)
The central task in many interactive machine learning systems can be formalized as the sequential optimization of a black-box function. Bayesian optimization (BO) is a powerful model-based framework for adaptive experimentation, where the primary goal is the optimization of the black-box function via sequentially chosen decisions. In many real-world tasks, it is essential for the decisions to be robust against, e.g., adversarial failures and perturbations, dynamic and time-varying phenomena, a mismatch between simulations and reality, etc. Under such requirements, the standard methods and BO algorithms become inadequate. In this talk, we discuss algorithms with provable regret guarantees that can enhance robust and adaptive decision making in BO and related problems. We also consider associated robust submodular and non-submodular optimization problems, and present practical and efficient algorithms with improved robustness and constant factor approximation guarantees. Finally, we demonstrate the robust performance of our algorithms in numerous real-world applications (e.g., environmental monitoring and recommender systems) and tasks (e.g., robot pushing and feature selection).
Key words: Bayesian optimization, Gaussian process, Submodularity, Robust optimization, Regret bounds, Level-set estimation, Non-submodular optimization
About the speaker — Volkan Cevher received the B.Sc. (valedictorian) in electrical engineering from Bilkent University in Ankara, Turkey, in 1999 and the Ph.D. in electrical and computer engineering from the Georgia Institute of Technology in Atlanta, GA in 2005. He was a Research Scientist with the University of Maryland, College Park from 2006-2007 and also with Rice University in Houston, TX, from 2008-2009. Currently, he is an Associate Professor at the Swiss Federal Institute of Technology Lausanne and a Faculty Fellow in the Electrical and Computer Engineering Department at Rice University. His research interests include signal processing theory, machine learning, convex optimization, and information theory. Dr. Cevher was the recipient of the IEEE Signal Processing Society Best Paper Award in 2016, a Best Paper Award at CAMSAP in 2015, a Best Paper Award at SPARS in 2009, and an ERC CG in 2016 as well as an ERC StG in 2011.
January 30, 11:00 — MED 2 1124 (CoViz2), EPFL
Development and validation of interatomic potentials and application to the simulation of phase transformations
Ralf Drautz (ICAMS, Ruhr-Universität Bochum)
I will present five short stories on the derivation of models of the interatomic interaction from Density Functional Theory (DFT), the validation of potentials and their application to the simulation of phase transformations. I will show how a systematic coarse graining of the electronic structure from DFT to the tight-binding approximation and analytic Bond-Order Potentials leads to magnetic potentials that capture the subtle interplay between magnetism and phase stability in iron. Further I will introduce the atomic cluster expansion as a formal many-atom expansion with an accuracy and transferability comparable to current machine learning approaches As different researchers typically have a different focus when developing potentials, interatomic potentials have various application ranges. I will present automated high-throughput calculations to validate a large number of interatomic potentials against DFT and to discern their application ranges I will then discuss the application of magnetic Bond-Order Potentials to simulating finite temperature magnetism in iron, in particular the ferromagnetic to paramagnetic phase transformation, the alpha-gamma transition and the prediction of mechanical properties. I will further summarize atomic simulations for phase stability, nucleation and solid-solid transformations with relevance to high-temperature materials.
About the speaker — Ralf Drautz received his Diploma in Physics (with distinction) from the Universität Stuttgart, Germany in 1998. He was a PhD student at the Max-Planck-Institut für Metallforschung, Stuttgart, Germany and received his PhD degree (Dr. rer. nat., summa cum laude) in 2003. Ralf Drautz was a research fellow (2003-2004), and a Senior Research Fellow and Materials Modelling Laboratory Research Fellow (2005-2008) at the University of Oxford. Since 2008, he has been Chair Professor at Ruhr-Universität Bochum in the Department of Physics and Astronomy, as well as the Director at the Interdisciplinary Centre for Advanced Materials Simulation (ICAMS).
Low-volume newsletters, targeted to the scientific and industrial communities.
Subscribe to our newsletter